Research
Our group develops computational methods for understanding the interactions, dynamics and conservation of complex biological systems. Below we highlight some of the problems we have worked on and some of our ongoing work.
Please see our publications page for complete details on these projects.
People








Identifying and modeling interactions
We have so far focused on two types of molecular interactions: Protein-DNA and protein protein. For protein-DNA we have developed methods for combining gene expression and CHIP-chip data for modeling regulatory networks in yeast. We are now extending these methods for modeling the dynamics of regulatory networks. Our work led to a number of new insights regarding the organization and combinatorial regulation of various sub-networks in yeast. For protein-protein interactions we are developing new method to extract the set of interacting proteins from high throughput biological datasets. We develop and test classification methods for this task and evaluate the importance of the different feature. The results of this work lead to many new hypothesis regarding the set of interacting proteins, some are currently tested by our collaborators.
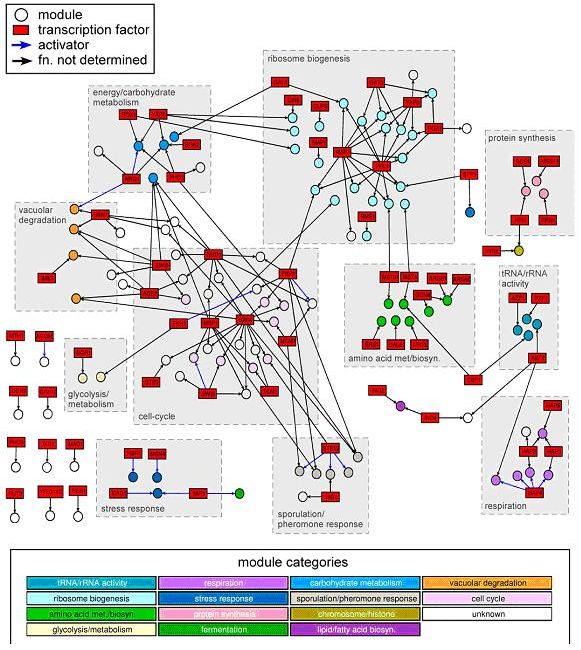
Figure 1: Rich media gene modules network.
Computational Discovery of Gene Modules and Regulatory Networks
Dynamics
We have primarily focused on the analysis and interpretation of time series gene expression data. Almost 40% of current gene expression datasets are time series and this percentage is likely to grow since most biological systems are dynamic. We have developed computational algorithms and tools for many different aspects of time series expression analysis including:
- Methods for designing time series experiments, focusing on when to sample and on the quality of time series expression profiles
- Methods for data analysis including mixed effects models for continuous representation of time series expression data, alignment and identifying differentially expressed genes in time series experiments. We have also developed methods for analyzing data from clinical expression experiments.
- Methods for patterns recognition in time series expression experiments. These include methods for optimal leaf ordering of hierarchical clustering results, for continuous clustering and for clustering short time series expression data.
- Methods for combining high throughput biological datasets for inferring regulatory networks in the cell. Many of the computational software and tools we have developed are available from the Software page.
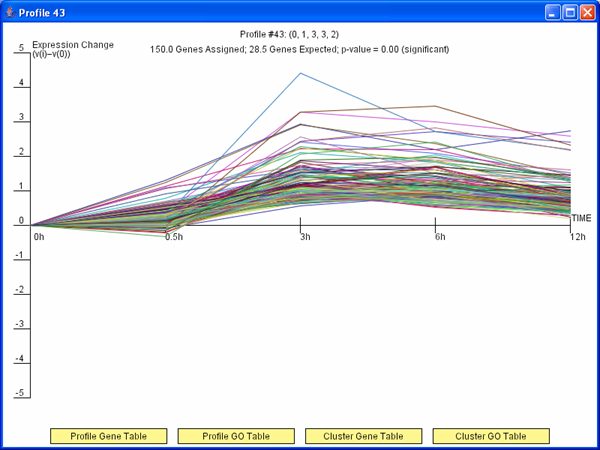
Figure 3: TODO
Short Time-series Expression Miner (STEM)
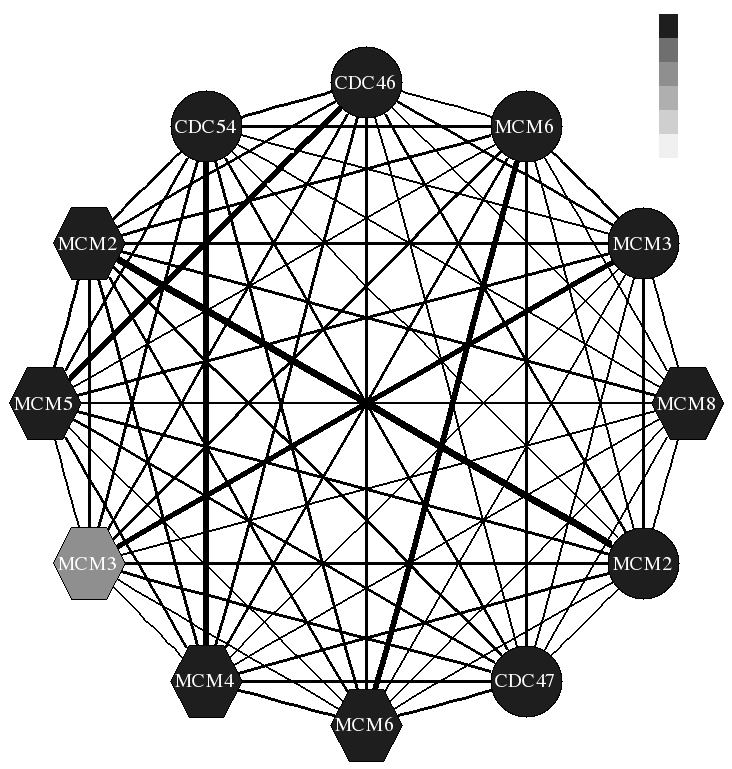
Conservation of biological systems
We have recently initiated a project that tries to combine (time series) expression data with sequence data to identify cell cycle genes. Our methods combine data from multiple species for this task allowing as improve upon the set discovered when using only the species data.